Article
Preparing for the future
The new European Union medical devices regulation
New regulations will impact all device manufacturers, so what are the steps manufacturers need to take to mitigate this impact? Given the scale and complexity associated with implementing the EU MDR changes it is important for manufacturers to adopt a structured enterprise wide cross-functional approach.
This report, developed by the UK Centre for Health Solutions, examines the proposals and impacts of regulation on medical devices.
Overview of the report
Since the 1990s, regulation of the medical device industry in Europe has been relatively unchanged. However, recent incidents have now prompted urgent regulatory and compliance reforms to the industry. Among the most significant of these are the European Commission’s 2012 proposals for regulation on medical devices (EU MDR) and in-vitro diagnostics (EU IVDR). With the formal publication of guidance imminent, the proposals will give national regulators much more control and oversight of the medical devices industry—with adoption mandatory. If companies do not comply with these upcoming changes then this could possibly result in a company losing its license to operate.
The impact of this regulation can dramatically alter the operations of medical device manufacturers and even impact the composition of their existing as well as future portfolios. Cost of compliance will most likely be significant. It is critical that businesses take action now—to gain stakeholder buy-in, prepare their organizations, and start implementing changes.
What are the impacts the EU MDR will have on the industry—from change management to portfolio reviews to product labelling? We propose a series of steps manufacturers should take to address and mitigate the changes ahead.
Step 1 - Understanding the EU MDR and IVDR
As a medical device manufacturer, importer or distributor it will be critical to have a good understanding of the new regulations, the scope and full impact on the business. Many companies will have combination products and so both the changing pharmaceutical and medical devices regulations are relevant. Understanding the overlap and synergies with other applicable regulations and directives such as IVD, Clinical Trial Regulation (CTR) for human use, Falsified Medicines Act and Identification of Medicinal Product (IDMP) will be important.
Step 2 - Medical device portfolio review and assessment
A manufacturer’s portfolio of products will need to be fully reviewed and assessed against the new regulations and future requirements. For example, under the new directive, products that are classified as accessories could now be covered under the definition of a medical device. With the new requirements there also may be product lines for which the classification status will change or the oversight by the Notified Bodies will be heightened without an increase in classification. It will be important to understand whether these products will need to be up-classified in the future and the associated impact. Some of the most critical areas that will require assessment include financial:
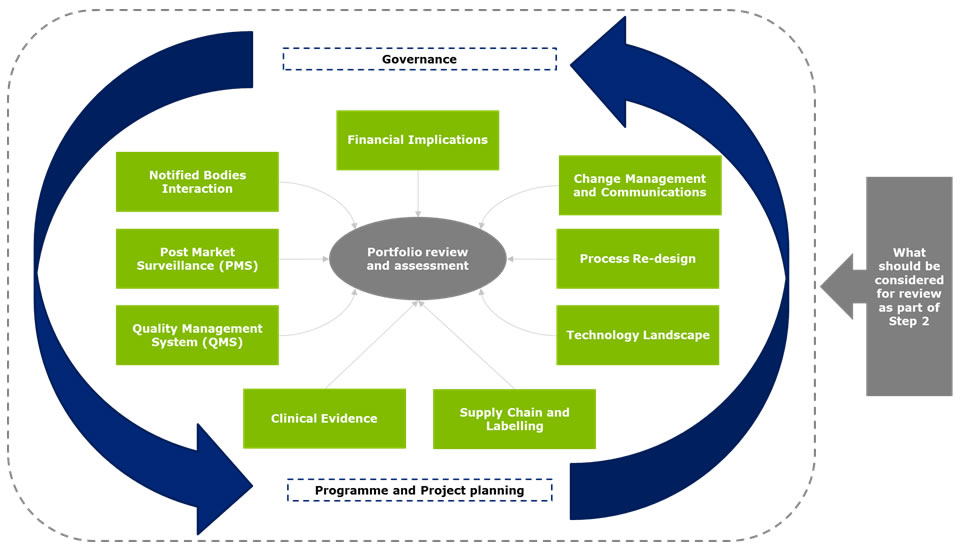
Step 3 – The EU MDR strategy and roadmap
Once a full portfolio review and assessment has been performed around the current and future states, the gaps can be defined. These gaps would essentially be classified as strategical and tactical projects, prioritized based on business, legal, and regulatory drivers.
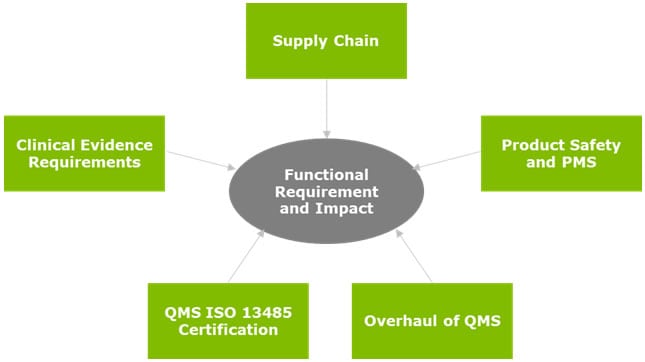
Conclusion
The level of time and effort needed for defining the strategy and planning for implementation of these regulations is not to be underestimated. In order to assess the impact of these changes on the business and its commercial and R&D operating models, organisations will need to build a robust business case and strong project management capabilities with effective cross-functional stakeholder management.
Contacts

Auch interessant

Identification of Medicinal Products
Think Transformation