医療機器のサイバーセキュリティ規制~デジタルヘルスも対象に? ブックマークが追加されました
概要
昨今の医療・ヘルスケア業界では、デジタル技術を駆使した価値提供がますます進んでおり、AI・IoT等を活用したデジタルヘルスの取組みが注目を集めています。日本でも医療機器におけるサイバーセキュリティ規制の運用が本格化しており、2023年4月付けで、厚生労働省が定める製造販売承認の基準が一部改正されました。※1
本稿では、医療機器の製造販売ビジネスを行うハードウェア開発事業者だけでなく、デジタルヘルスのソフトウェア開発を手掛ける事業者や、新たに医療・ヘルスケア業界に参入する事業者を対象に、サイバーセキュリティの観点から必要な対策についてお伝えします。
① はじめに(医療機器の規制とは?)
はじめに、医療機器を製造販売する際には、品質・有効性・安全性の面で問題がないことを審査し、「薬事承認」を取得しなければならないという規制があります。この薬事承認の基準「基本要件基準」は、法律(通称、医薬品医療機器等法)で定められています。
薬事承認申請後は、「PMDA(独立行政法人 医薬品医療機器総合機構)による審査→厚生労働省による承認→保険適用の手続き」が行われます。
なお、医療機器のクラス分類によっては、厚生労働省の承認の代わりに、民間の登録認証機関による第三者「認証」の取得や、「届出」等の対応をする場合もあります。
「基本要件基準」改正によるビジネスへの影響
今回、2023年4月に改正された「基本要件基準」において、サイバーセキュリティの確保に関する要求事項が追加されました。これにより、今後、医療機器の薬事承認を取得する際は、サイバーセキュリティへの対応が求められます。2023年3月末までは経過措置として従前の例によることができるものの、2024年4月以降はサイバーセキュリティ対応状況の確認が必須化されます。
改正の背景
図1に示すとおり、各国の医療機器規制を取りまとめている国際フォーラムIMDRF(International Medical Device Regulators Forum)が2020年および2023年にサイバーセキュリティのガイダンスを公開しました。※2,3,4 この背景には、近年の医療機器製品の国際的な流通量の増加と、インターネットに接続される医療機器の増加により、医療機器が国境を越えてサイバー攻撃を受ける可能性が高まっていることが挙げられます。
これを踏まえ日本も、2021年12月に厚生労働省より、IMDRFの国際的なベストプラクティスを採用した「医療機器のサイバーセキュリティ導入に関する手引書(以下、手引書)」が公開され※5、2023年4月に規制が改正されました。
今回の規制改正により、サイバーセキュリティの確保に関する要件(後述)を満たしていなければ、日本国内では製品の製造販売の承認が得られず、ビジネスをできなくなる可能性があります。
図1:医療機器関連規制の国内外における動向

② 規制改正の影響を受ける対象
今回規定された基本要件基準第12条3項では、規制の影響範囲として、「プログラムを用いた医療機器」への対応が求められることが明記されています。とりわけ、他の機器やネットワークに接続して使用される医療機器や、外部からの不正アクセスや攻撃を受ける可能性のあるソフトウェアが規制の対象となります。
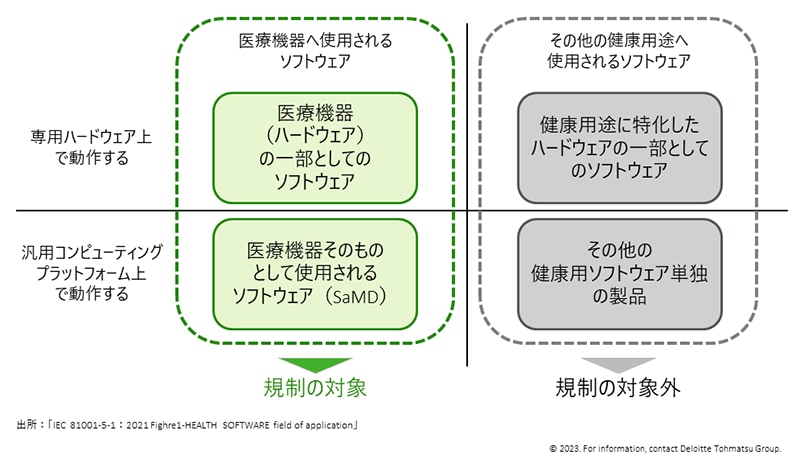
つまり、図2に示すように、医療機器のハードウェアに組み込まれたソフトウェアだけでなく、治療用アプリ等、汎用コンピュータやスマートフォン等の携帯情報端末にインストールして医療機器として使用されるソフトウェア(医療機器プログラム/SaMD;Software as a Medical Device)も規制の対象となり、関連する幅広い事業者が今回の規制改正の影響を受けることとなります。
なお、厚生労働省の判断事例によると、診断や治療等を目的としない、個人利用の健康管理ソフトウェア等は医療機器には該当しないため、今回の規制の対象外と考えられます。※6 医療機器プログラムの基本的な考え方としては、「医療機器としての目的性を有しており、かつ、プログラムが意図したとおりに機能しない場合に生命や健康に影響を与えるおそれがあるプログラム」が医療機器に該当しますが、対象となるプログラムの考え方や規制範囲については今後変更が生じる可能性があります。※7
図2:改正された医療機器規制の対象となるソフトウェア
③ 対応のポイント
基本要件基準第12条3項への対応は、2023年4月1日から適用され、1年間の経過措置期間が設けられます。したがって、製品の申請タイミングによって対応が異なります。※8
改正に伴う対応時期の考え方
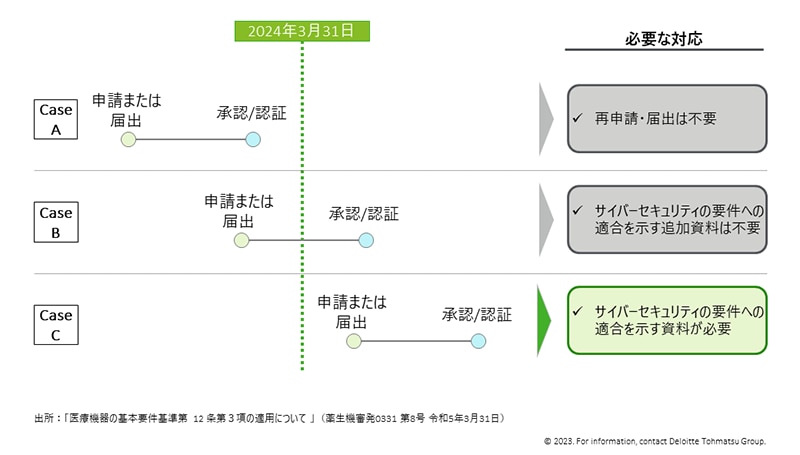
基本要件基準の改正に伴う対応時期の考え方は、図3に示すとおり、2024年3月31日を境界にして3通りのケースが考えられます。※8
Case A) 2024年3月31日以前に承認/認証の申請または届出が完了している場合
改正による再申請・届出は不要
Case B) 2024年3月31日以前に承認/認証の申請または届出を行う場合
改正による再申請・届出は不要
Case C) 2024年4月1日以降に承認/認証の申請または届出を行う場合
改正された基本要件基準への適合確認と、それを示す資料の提出が必要
図3:改正に伴う対応時期の考え方
【補足】既に申請済み、上市済みの医療機器への対応
既に申請済みの場合は、Case AやCase Bに該当し、前述のとおり再度の申請や届出は必要ありません。ただし、上市後における医療機器の機能拡大など、有効性及び安全性に影響を与えるおそれのある変更等が発生する場合は、「一部変更申請」として位置づけられ、2024年4月1日以降の一部変更申請では、改正された基本要件基準への適合確認と、必要に応じて資料の提出が必要です。※8 既に製造販売されている医療機器の取扱いについては、厚生労働省から通知される予定です。※8
対応すべきサイバーセキュリティの確保に関する要件
新設された基本要件基準第12条第3項への対応としては、製品ライフサイクル全体におけるサイバーセキュリティの取組みを規定した規格等への適合性の確認が必要になります。具体的には、ヘルスソフトウェア向けの規格であるJIS T 81001-5-1や、IMDRFガイダンスをベースとした厚生労働省の手引書を参照することができます。
JIS T 81001-5-1は、製造販売業者が従来から求められているリスクマネジメントの要求事項に加えて、サイバーセキュリティに関する取組みを規定した規格です。例えば、設計・開発時の脅威モデリングやリスク分析、セキュリティテスト、保守のための構成管理、問題発生時の対応等が含まれます。
また手引書には、製品サイバーセキュリティの取組みの前提として、下記が必要であると述べられています。※5
・製品のセキュリティポリシー設定
・セキュリティの定量的評価、反復試験、侵入試験等の能力向上
・ PSIRT(Product Security Incident Response Team)等の製品セキュリティ体制の構築
・一連のサイバーセキュリティのベースラインとなる活動を定め、品質マネジメントシステム(QMS)の中に定着させる取組み
これらの要求事項を基に、組織内でサイバーセキュリティを確保する方針を定め、運用プロセスを整備し、組織全体に浸透させる必要があります。そのために重要なことは、「製品セキュリティの取組み全体を統制する部署」を設置することです。組織内に、製品開発のプロジェクトに依存しない標準化されたサイバーセキュリティ品質管理の仕組みが存在することで、円滑にリスク管理をすることができます。
特に、医療機器メーカー等の製造業では、製品セキュリティ品質を統制する専門部署を設置していることが多い一方、デジタルヘルスのソフトウェア開発を主事業とする企業では、十分な体制が整っていない場合もあります。そのため、サイバーセキュリティを製品品質の一部と捉え、確実な取組みを推進するために、組織内での役割分担整理や体制整備が必要です。
④ まとめ
2024年4月1日までの経過措置期間は設けられているものの、製品セキュリティポリシーやPSIRT等の基盤を整え、組織内にサイバーセキュリティ要件を浸透させるには、相当な時間を要します。新規事業としてデジタルヘルスの開発に取り組む企業では、製品の品質・安全性・有効性を確保する管理体制をゼロから構築するケースも多いことから、薬事申請をスムーズに進めるためには、不足している体制やプロセスを洗い出す等、上市に向けた準備を着実に進めることが重要です。
≪参考文献≫
・※1: 国立印刷局,「厚生労働省告示第六十七号」 官報 第933号,(発行 2023年3月9日(木))
・※2: IMDRF/CYBER WG/N60, ”Principles and Practices for Medical Device Cybersecurity”(発行2020-04-20)
・※3: IMDRF/CYBER WG/N70, ”Principles and Practices for the Cybersecurity of Legacy Medical Devices”(発行2023-04-11)
・※4: IMDRF/CYBER WG/N73, ”Principles and Practices for Software Bill of Materials for Medical Device Cybersecurity”(発行2023-04-13)
・※5: 日本医療機器産業連合会 医療機器サイバーセキュリティ対応WG,「医療機器のサイバーセキュリティ導入に関する手引書(第2版)」(発行2023-03-31)
・※6: 厚生労働省,「プログラムの医療機器該当性判断事例について」(発行2023-3-31)
・※7: 厚生労働省,「プログラムの医療機器該当性に関するガイドライン」(一部改正 2023-3-31)
・※8: 厚生労働省,「医療機器の基本要件基準第 12 条第3項の適用について」薬生機審発0331 第8号, (発行2023-3-31)
≪執筆者≫
岩本 高明、北市 恭奨、菅沼 優里
執筆者
北市 恭奨/Kyosuke Kitaichi
デロイト トーマツ サイバー合同会社
デロイト トーマツ コンサルティングに入社後、経済安全保障×サイバーセキュリティを主軸に活動。2019年よりデロイト トーマツ サイバーに参画。ライフサイエンス・ヘルスケア業界を中心に、サイバー戦略策定やグローバルガバナンス態勢構築、組織風土変革、セキュリティを起点とした製品・サービスの事業戦略およびルール形成戦略策定など、サイバーストラテジー領域の幅広い経験を有する。
菅沼 優里/Yuri Suganuma
デロイト トーマツ サイバー合同会社
医療介護機器メーカーを経て、2022年にデロイト トーマツ サイバー合同会社へ入社。一貫してサイバーセキュリティ戦略立案に従事し、セキュリティポリシー・ルールの策定、組織体制の見直し、製品上市に向けたセキュリティ戦略ロードマップ策定等のリスクアドバイザリー経験を有する。
※所属などの情報は執筆当時のものです。
プロフェッショナル

岩本 高明/Takaaki Iwamoto
デロイト トーマツ サイバー合同会社 執行役員
大手インテグレーター、戦略系コンサルファームを経て現職。企業に対するサイバーセキュリティ戦略立案、リスク分析・対応方針立案等の業務を歴任。CISO等の経営アジェンダを広くカバーする一方、技術対策までサイバー全体に一貫整合した経験を有する。
Recommended for you
Opens_in_a_new_window